
In: Handbook ground water; volume II, methodology
Environmental Protection Agency
Introduction
Protection and remediation of ground-water resources require an understanding of processes that affect fate and transport of contaminants in the subsurface environment. This understanding allows: (1) prediction of the time of arrival and concentration of contaminants at a receptor, such as a monitoring well, a water supply well, or a body of surface water; (2) design of costeffective and safe waste management facilities; (3) installation of effective monitoring systems; and (4) development of efficient and cost-effective strategies for remediation of contaminated aquifers (Palmer and Johnson, 1989a).
Contaminants in ground water will move primarily in a horizontal direction that is determined by the hydraulic gradient. The contaminants will decrease in concentration because of such processes as dispersion (molecular and hydrodynamic), filtration, sorption, various chemical processes, microbial degradation, time rate release of contaminants, and distance of travel (U.S. Environmental Protection Agency, 1985). Processes such as hydrodynamic dispersion affect all contaminants equally, while sorption, chemical processes, and degradation may affect various contaminants at different rates. The complex factors that control the movement of contaminants in ground water and the resulting behavior of contaminant plumes are commonly difficult to assess because of the interaction of the many factors that affect the extent and rate of contaminant movement. Predictions of movement and behavior can be used only as estimates, and modeling is often a useful tool to integrate the various factors.
The U.S. Environmental Protection Agency (EPA) sponsored a series of technology transfer seminars between October 1987 and February 1988 that provided an overview of the physical, chemical, and biological processes that govern the transport and fate of contaminants in the subsurface. The following discussion is a summary of the workshops, and is based on the seminar publication, Transport and Fate of Contaminants in the Subsurface (U.S. Environmental Protection Agency, 1989).
Physical Processes Controlling the Transport of Contaminants in the Aqueous Phase in the Subsurface
Advection-Dispersion Theory
The study of advection and dispersion processes is useful for predicting the time when an action limit, i.e., a concentration limit used in regulations such as drinking water standards, will be reached. Knowledge of advection-dispersion also can be used to select technically accurate and cost-effective remedial technologies for contaminated aquifers.
If concentrations of a contaminant were measured in a monitoring well that was located between a contaminant source and a receptor such as a water supply well, a graph of concentrations versus time would show a breakthrough curve, i.e., the concentrations do not increase in a step-function (i.e., plug flow), but rather in an S-shaped curve (Figure 3-1). In a one-dimensional, homogeneous system, the arrival of the center of the mass is due to advection, while the spread of the breakthrough curve is the result of dispersion (Palmer and Johnson, 1989a).

Figure 3-1. Breakthrough Curve for a Contaminant, as Measured in a Monitoring Well (Palmer and Johnson, 1989a).
Advection
Advection is defined by the transport of a non-reactive, conservative tracer at an average ground-watervelocity (Palmer and Johnson, 1989a). The average linear velocity is dependent on (1) the hydraulic conductivity of the subsurface geologic formation in the direction of ground-water flow, (2) the porosity of the formation and (3) the hydraulic gradient in the direction of groundwater flow. For waste contaminants that react through precipitation/dissolution, adsorption, and/or partitioning reactions within the subsurface formation, the velocity can be different from the average ground-water velocity.
Dispersion
Dispersion of waste contaminants in an aquifer causes the concentration of contaminants to decrease with increasing length of flow (U.S. Environmental Protection Agency, 1985). Dispersion is caused by: (1) molecular diffusion (important only at very low velocities) and (2) hydrodynamic mixing (occurring at higher velocities in laminar flow through porous media). Contaminants traveling through porous media have different velocities and flow paths with different lengths. Contaminants moving along a shorter flow path or at a higher velocity, therefore, arrive at a specific point sooner than contaminants following a longer path or traveling at a lower velocity, resulting in hydrodynamic dispersion.
Figure 3-2 shows that dispersion can occur in both longitudinal (in the direction of ground-water flow) and transverse (perpendicular to ground-water flow) directions, resulting in the formation of a conic waste plume downstream from a continuous pollution source (U.S. Environmental Protection Agency, 1985). The concentration of waste contaminants is less at the margins of the plume and increases towards the source. A plume will increase in size with more rapid flow within a time period, because dispersion is directly related to ground-water velocity.
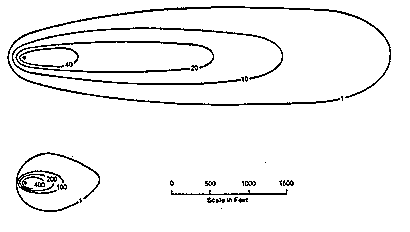
Figure 3-2 The Effects of Ground-Water Velocity on Plume Shape. Upper Plume Velocity: 1.5 ft/day and Lower Plume Velocity: 0.5 ft/day (US.Environmental Protection Agency, 1985).
The dispersion coefficient varies with ground-water velocity. At low velocity, the dispersion coefficient is relatively constant, but increases linearly with velocity as ground-water velocity increases. Based on these observations, investigators proposed that the dispersion coefficient can be expressed as a sum of an effective molecular diffusion coefficient and a mechanical dispersion coefficient (Palmer and Johnson, 1989a).
The effective molecular diffusion coefficient is a function of the solution diffusion coefficient and the tortuosity of the medium. Tortuosity accounts for the increased distance a diffusing ion must travel around sand grains. The mechanical dispersion coefficient is proportional to velocity. Specifically, mechanical dispersion is a result of: (1) velocity variations within a pore, (2) different pore geometries, and (3) divergence of flow lines around sand grains present in a porous medium (Gillham and Cherry, 1982).
The term dispersivity is often confused with dispersion. Dispersivity does not include velocity, so to convert dispersivity to dispersion requires multiplication by velocity. Since dispersion is dependent on site-specific velocity parameters and configuration of pore spaces within an aquifer, a dispersion coefficient should be determined experimentally or empirically for a specific aquifer. The selection of appropriate dispersion coefficients that adequately reflect existing aquifer conditions is critical to the success of chemical transport modeling (U.S. Environmental Protection Agency, 1985).
Advection-Dispersion Equation
An advection-dispersion equation is used to express the mass balance of a waste contaminant within an aquifer as a result of dispersion, advection, and change in storage. The mass balance is a function of the dispersion coefficient, the ground-water velocity, concentration of the contaminant, distance, and time (Palmer andJohnson,1989a). An advection-dispersion equation can be applied to the description of threedimensional transport of waste contaminants in an aquifer, using three dispersion coefficients (one longitudinal and two transverse). Mathematically detailed descriptions of the advection-dispersion equation are presented in Bear (1969, 1979).
Discrepancies between results generated from advection-dispersion equations and laboratory and field experiments have been found. These discrepancies have been attributed to: (1) immobile zones of water withinthe aquifer, (2) solution-solid interface processes, (3) anion exclusion, and (4) diffusion in and out of aggregates (Palmer and Johnson, 1989a).
Field observations using field tracer studies also have shown that longitudinal dispersivity values are usually much larger than transverse dispersivity measurements (Palmer and Johnson, 1989a). Figure 3-3 shows three-dimensional field monitoring that has corroborated these observations by identifying long, thin contaminant plumes rather than plumes spread over the thickness of an aquifer. (Kimmel and Braids, 1980; MacFarlane and others, 1983). The large longitudinal dispersion coefficients are thought to result from aquifer heterogeneity. In an ideally stratified aquifer with layers of sediment of different hydraulic conductivities, contaminants move rapidly along layers with higher permeabilities and more slowly along the lower permeability layers (Figure 3-4) (Palmer and Johnson, 1989a). Sample concentration of a contaminant is an integration of the concentrations of each layer, if water is sampled from monitoring wells that are screened through the various layers.

Figure 3-3. Hypothetical Contaminant Plumes for Large (A) and Small (B) Dispersivities (Palmer and Johnson, 1989a).
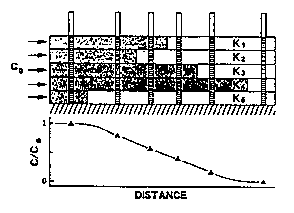
Figure 3-4. Contaminant Distributions and Concentrations In an Ideally Stratified Aquifer (after Gillham and Cherry, 1982, by Palmer and Johnson, 1989a).
Results from plotting concentration versus distance show a curve with large differences in concentrations, even though only advection is considered. This dispersion is the result of aquifer heterogeneity and not pore-scale processes.
However, defining hydraulic conductivities in the subsurface is difficult, since not all geologic formations are perfectly stratified, but may contain cross stratification or graded bedding (Palmer and Johnson, 1989a). To quantify heterogeneity in an aquifer, hydraulic conductivity is considered to be random, and statistical characteristics, such as mean, variance, and autocorrelation function, are determined.
In addition to aquifer heterogeneity, other processes, contributing to the spread of contaminants include: (1) diverging flow lines resulting in the spread of contaminants by advection over a larger cross section of the aquifer, (2) temporal variations in the water table resulting in change of direction of ground-water flow and lateral spread of contamination, and (3) variations in concentration of contaminants at the source resulting in apparent dispersion in the longitudinal direction (Frind .d Hokkanen, 1987; Palmer and Johnson, 1989a).
Ground-water sampling methods also may result in detection of apparent spreading of contaminant plumes (Palmer and Johnson, 1989a). An underestimation of contaminant concentrations at specific locations in an aquifer may be due to insufficient well-purging. Monitoring wells with different screen lengths that integrate ground water from different sections of the aquifer may yield dissimilar contaminant concentrations.
Diffusive Transport through Low Permeability Materials
In materials with low hydraulic conductivities (e.g., unfractured clays and rocks with conductivities less than 10 to 9 m/s), diffusive transport of waste contaminants is large compared to advective transport (Neuzil, 1986; Palmer and Johnson, 1989a). Contaminants can diffuse across natural aquitards or clay liners with low hydraulic conductivities, resulting in aquifer contamination. The extent of movement is dependent on diffusive flux, rate of ground-water flow in the aquifer, and the length of the source area in the direction of ground-water flow.
Effects of Density on Transport of Contaminants
The density of a contaminant plume may contribute to the direction of solute transport if dissolved concentrations of contaminants are large enough (Palmer and Johnson, 1989a). For example, assume that the density of ground water within an aquifer is 1.00, the natural horizontal gradient is 0.005, and the natural vertical gradient is 0.000. If the density of the contaminant plume is equal to the density of the ground water, the plume moves horizontally with the naturally existing hydraulic gradient. If the density of the contaminated water is 1.005 (a concentration of approximately 7,000 mg/L total dissolved solids), then the driving force in the vertical direction is the same as the driving force in the horizontal direction. If the aquifer is isotropic, then the resulting vector of these two forces descends at 45 degrees into the aquifer. The contaminant plume moves deeply into the aquifer and may not be detected with shallow monitoring systems installed under the assumption of horizontal flow.
Retardation of Contaminants
If contaminants undergo chemical reactions while being transported through an aquifer, their movement rate may be less than the average ground-water flow rate (Palmer and Johnson,1989a). Such chemical reactions that slow movement of contaminants in an aquifer include precipitation, adsorption, ion exchange, and partitioning into organic matter or organic solvents. Chemical reactions affect contaminant breakthrough, as shown in Figure 3-5. If the retardation factor, R (calculated from equations for contaminant transport that include retardation), is equal to 1.0, the solute is nonreactive and moves with the ground water. If R is greater than 1.0, the average velocity of the solute is less than the velocity of the ground water, and the dispersion of the solute is reduced. If a monitoring well is located a distance from a contaminant source such that a nonreactive solute requires time, t1, to travel from the source to the well, a contaminant with a retardation factor of 2 will require 2t1 to reach the well, and 4t1 will be required for a contaminant with a retardation factor of 4.
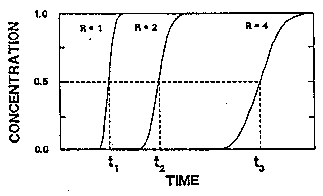
Figure 3-5. Time Required for Movement of Contaminants at Different Retardation Factors (Palmer and Johnson, 1989a).
Contaminants with lower retardation factors are transported greater distances over a given time than contaminants with larger retardation factors (Figure 3-6) (Palmer and Johnson, 1989a). A monitoring well network has a greater chance of detecting contaminants with lower retardation factors because they are found in a greater volume of the aquifer. Estimates of the total mass of a contaminant with a retardation factor of 1.0 in an aquifer may be more accurate than estimates for contaminants with greater amounts of retardation. Therefore, estimates of time required to remove nonreactive contaminants may be more accurate than time estimates for retarded contaminants. The slow movement of retarded contaminants may control the time and costs required to remediate a contaminated aquifer.
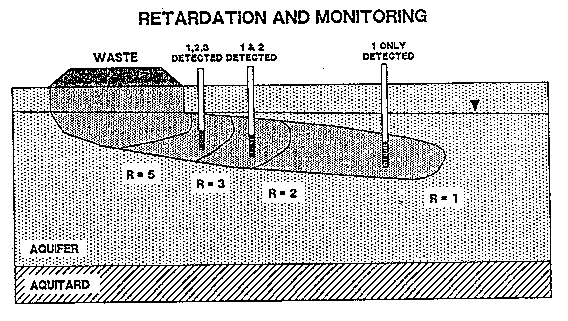
Figure 3-6. Transport of Contaminants with Varying Retardation Factors at a Waste Site (Palmer and Johnson, 1989a).
Transport through Fractured Media
Because fractured rock has both primary and secondary porosity, models used to describe solute transport in porous media, such as aquifers in recent alluvial deposits or glacial sediments, may not be appropriate for use at sites on fractured rock (Palmer and Johnson, 1989a). Primary porosity is the pore space formed at the time of deposition and formation of the rock mass, and secondary porosity is the pore space formed as the result of fracture of the rock.
Transport mechanisms in fractured media are advection and dispersion, the same as in porous media (Figure 3-7) (Palmer and Johnson, 1989a). In fractured media, however, contaminants are transported by advection only along fractures. Dispersion in fractured media is due to: (1) mixing at fracture intersections, (2) variations in opening widths across the width of the fracture, (3) variations in opening widths along stream lines, (4) molecular diffusion into microfractures penetrating the interfracture blocks and (5) molecular diffusion into interfracture porous matrix blocks (more important in fractured porous rock than in fractured crystalline rock).
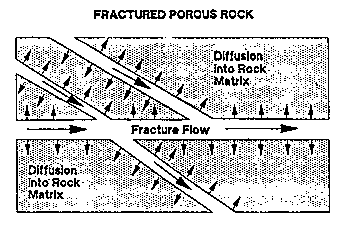
Figure 3-7. Transport in Fractured Porous Rock (Palmer and Johnson, 1989a).
Transport of contaminants through fractured media is described by one of four general models: continuum, discrete fracture, hybrid, and channel (Palmer and Johnson, 1989a).
In continuum models, individual fractures are ignored and the entire medium is considered to act as an equivalent porous medium. Single porosity continuum models are applicable where the only porosity of the rock mass is the fracture porosity, such as in fractured granite or basalt. Double porosity models are applicable to media in which there is both primary and secondary porosity such as sandstones and shales.
Discrete fracture models try to describe flow and transport in individual fractures. Because it can be difficult to obtain information about each fracture in the rock mass, stochastic models usually are required. These models use statistical information about distribution of fracture properties such as orientation and aperture widths to describe flow and transport.
Hybrid models are combinations of discrete fracture and continuum models, while channel models describe solute transport as small fingers or channels rather than as a uniform front along the width of a fracture.
Particle Transport through Porous Media
In addition to solute transport through porous media, the transport of particles (including bacteria, viruses, inorganic precipitates, natural organic matter, asbestos fibers, or clays) also may be important in investigations of contaminant transport. Particles can be removed from solution by surface filtration, straining, and physical-chemical processes (Figure 3-8) (Palmer and Johnson, 1989a).
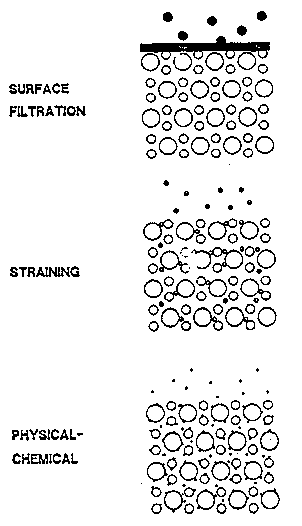
Figure 3-8. Mechanisms of Filtration (Palmer and Johnson, 1989a).
The effectiveness of each process is dependent on the size of the specific particles present (Palmer and Johnson, 1989a). If particles are larger than the largest pore diameters, they cannot penetrate into the porous medium and are filtered at the surface of the medium. If particles are smaller than the largest pores but larger than the smallest, the particles are transported through the larger pore channels, but eventually encounter a pore channel with a smaller diameter and are removed by straining. If particles are smaller than the smallest pore openings, the particles can be transported long distances through the porous medium.
The rate at which particles move through the porous medium depends on several physical-chemical processes (Palmer and Johnson, 1989a). Particles may undergo random collisions with sand grains, and in a percentage of those collisions particles will adhere to the solid matrix by interception. Chemical conditions may affect particle transport; e.g., such processes as aggregation formation due to pH changes may change particle surface properties. These larger aggregates can then be strained or filtered from the water.
Microorganism movement through geologic materials is limited by many processes (Palmer and Johnson, 1989a). Some bacteria are large enough to be strained from the water. Although viruses, which are smaller than bacteria, can pass through the pores, they may adsorb to geologic materials because their surfaces are charged. Microorganisms, like chemical constituents, can be transported by diffusion, or if they are motile, can move in response to changes in environmental conditions and chemical concentrations. Since microorganism live and die, the rates of these processes should be included in the description of their transport in the subsurface.
Physical Processes Controlling the Transport of Non-Aqueous Phase Liquids (NAPLs) in the Subsurface
Transport and Dissolution of NAPLs
Non-aqueous phase liquids (NAPLs) are those liquids that do not readily dissolve in water and can exist as a separate fluid phase. (Palmer and Johnson, 1989b). NAPLs are divided into two classes: those that are lighter than water (LNAPLs) and those with a density greater than water (DNAPLs). LNAPLs include hydrocarbon fuels, such as gasoline, heating oil, kerosene, jet fuel, and aviation gas. DNAPLs include the chlorinated hydrocarbons, such as 1,1,1-trichloroethane, carbon tetrachloride, chlorophenols, chlorobenzenes, tetrachloroethylene, and polychlorinated biphenyls (PCBs).
As NAPLs move through geologic media, they displace water and air (Palmer and Johnson, 1989b). Water is the wetting phase relative to both air and NAPLs and tends to line edges of pores and cover sand grains. NAPLs are the non-wetting phase and tend to move through the center of pore spaces. Neither the water nor the NAPL phase occupies the entire pore, so the permeability of the medium with respect to these fluids is different than when the pore space is entirely occupied by a single phase. This reduction in permeability depends upon the specific medium and can be described in terms of relative permeability, i.e., permeability at a certain fraction of pore space occupied by the NAPL compared to the permeability of the medium at saturation with the NAPL. Relative permeability ranges from 1.0 at 100 percent saturation to 0.0 at 0 percent saturation.
Figure 3-9 shows permeabilityof a NAPL in a hypothetical medium during multiphase flow. (Palmer and Johnson, 1989b). At 100 percent water saturation, the relative permeabilities of the water and NAPL are 1.0 and 0.0, respectively. As the fraction of the pore space occupied by NAPL increases, a corresponding decrease occurs in the fraction of water within the pore space. As the water fraction decreases, the relative permeability with respect to the water phase decreases to zero. Zero relative permeability is not obtained when the fraction of water within the pore space equals zero, but at the irreducible water saturation (Srw), i.e., the level of water saturation at which the water phase is effectively immobile and there is no significant flow of water. The relative permeability of NAPL is similar. At 100 percent NAPL saturation, the relative permeability for the NAPL is equal to 1.0, but as the NAPL saturation decreases, the relative permeability of the NAPL decreases. At the residual NAPL saturation (Srn), the relative permeability for the NAPL is effectively zero, and the NAPL is considered immobile. These immobile fractions of NAPL cannot be easily removed from pores except by dissolution by flowing water.

Figure 3-9. Relative Permeability as a Function of Saturation (Palmer and Johnson, 1989b).
Transport of Light NAPLs
If small volumes of a spilled LNAPL enter the unsaturated zone (i.e., vadose zone), the LNAPL will flow through the central portion of the unsaturated pores until residual saturation is reached (Figure 3-10a) (Palmer and Johnson, 1989b). A three-phase system consisting of water, LNAPL, and air is formed within the vadose zone. Infiltrating water dissolves the components within the LNAPL (e.g., benzene, xylene, and toluene) and transports them to the water table. These dissolved contaminants form a contaminated plume radiating from the area of the residual product. Many components found in LNAPLs are volatile and can partition into soil air and be transported by molecular diffusion to other parts of the aquifer. As these vapors diffuse into adjoining soil areas, they may partition back into the water phase and transfer contamination over wider areas. If the soil surface is relatively impermeable, vapors will not diffuse across the surface boundary and concentrations of contaminants in the soil atmosphere may build up to equilibrium conditions. However, if the surface is not covered with an impermeable material, vapors may diffuse into the atmosphere.
If large volumes of LNAPL are spilled (Figure 3-10b), the LNAPL flows through the pore space to the top of the capillary fringe of the water table. Dissolved components of the LNAPL precede the less soluble components and may change the wetting properties of the water, causing a reduction in the residual water content and a decrease in the height of the capillary fringe.
Since LNAPLs are lighter than water, they will float on top of the capillary fringe. As the head formed by the infiltrating LNAPLs increases, the water table is depressed and the LNAPLs accumulate in the depression. If the source of the spilled LNAPLs is removed or contained; LNAPLs within the vadose zone continue to flow under the force of gravity until reaching residual saturation. As the LNAPLs continue to enter the water table depression, they spread laterally on top of the capillary fringe (Figure 3-10c). The draining of the upper portions of the vadose zone reduces the total head at the interface between the LNAPLs and the ground water, causing the water table to rebound slightly. The rebounding water displaces only a portion of the LNAPLs because the LNAPLs remain at residual saturation. Ground water passing through the area of residual saturation dissolves constituents of the residual LNAPLs, forming a contaminant plume. Water infiltrating from the surface also can dissolve the residual LNAPLs and add to the contaminant load of the aquifer.
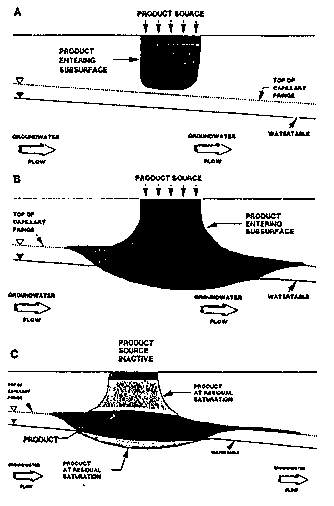
Figure 3-10. Movement of LNAPLs into the Subsurface: (A) Distribution of LNAPLs after Small Volume has Been Spilled; (B) Depression of the Capillary Fringe and Water Table; (C) Rebounding of the Water Table as LNAPLs Drain From Overlying Pore Space (Palmer and Johnson, 1989b).
Decrease in the water table level from seasonal variations or ground-water pumping also causes dropping of the pool of LNAPLs. If the water table rises again, part of the LNAPLs may be pushed up, but a portion remains at residual saturation below the new water table. Variations in the water table height, therefore, can spread LNAPLs over a greater thickness of the aquifer, causing larger volumes of aquifer materials to be contaminated. Selection of a remedial technology for LNAPLs in the ground water should not include techniques that move LNAPLs into uncontaminated areas where more LNAPLs can be held at residual saturation.
Transport of Dense NAPLs
DNAPLs are very mobile in the subsurface because of their relatively low solubility, high density, and low viscosity (Palmer and Johnson, 1989b). The low solubility means that DNAPLs do not readily mix with water and remain as separate phases. Their high density provides a driving force that can carry them deep into aquifers.
The combination of high density and low viscosity results in the displacement of the lower density, higher viscosity fluid, i.e., water, by DNAPLs, causing “unstable” flow and viscous fingering (Saffman and Taylor, 1958; Chouke and others, 1959; Homsy, 1987; Kueper and Frind, 1988).
If a small amount of DNAPL is spilled (Figure 3-11a), the DNAPL will flow through the unsaturated zone under the influence of gravity toward the water table, flowing until reaching residual saturation in the unsaturated zone (Palmer and Johnson, 1989b). If water is present in the vadose zone, viscous fingering of the DNAPLs will be observed during infiltration. No viscous fingering will be exhibited if the unsaturated zone is dry. The DNAPLs can partition into the vapor phase, with the dense vapors sinking to the capillary fringe. Residual DNAPLs or vapors can be dissolved by infiltrating water and be transported to the water table, resulting in a contaminant plume within the aquifer.
If a greater amount of DNAPL is spilled (Figure 3-11 b), the DNAPLs flow until they reach the capillary fringe and begin to penetrate the aquifer. To move through the capillary fringe, the DNAPLs must overcome the capillary forces between the water and the medium. A critical height of DNAPLs is required to overcome these forces. Larger critical heights are required for DNAPLs to move through unfractured, saturated clays and silts; thus these types of materials may be effective barriers to the movement of DNAPLs if the critical heights are not exceeded.
After penetrating the aquifer, DNAPLs continue to move through the saturated zone until they reach residual saturation. DNAPLs are then dissolved by ground water passing through the contaminated area, resulting in a contaminant plume that can extend over a large thickness of the aquifer. If finer-grained strata are contained within the aquifer, infiltrating DNAPLs accumulate on top of the strata, creating a pool. At the interface between the ground water and the DNAPL pool, the solvent dissolves into the water and spreads vertically by molecular diffusion. As water flows by the DNAPL pool, the concentration of the contaminants in the ground water increases until saturation is achieved or the downgradient edge of the pool is reached. DNAPLs, therefore, often exist in fingers or pools in the subsurface, rather than in continuous distributions. The density of pools and fingers of DNAPLs within an aquifer are important for controlling the concentrations of dissolved contaminants originating from DNAPLs.
If even larger amounts of DNAPLs are spilled (Figure 311c), DNAPLs can penetrate to the bottom of the aquifer, forming pools in depressions. If the impermeable lower boundary is sloping, DNAPLs flow down the dip of the boundary. This direction can be upgradient from the original spill area if the impermeable boundary slopes in that direction. DNAPLs also can flow along bedrock troughs, which may be oriented differently from the direction of ground-water flow. Flow along impermeable boundaries can spread contamination in directions that would not be predicted based on hydraulics.

Figure 3-11. Movement of DNAPLs into the Subsurface (A) Distribution of DNAPLs after Small Volume has Been Spilled; (B) Distribution of DNAPLs after Moderate Volume has Been Spilled; (C) Distribution of DNAPLs after Large Volume has Been Spilled (after Feenstra and Cherry, 1988, by Palmer and Johnson, 1989b).
Chemical Processes Controlling the Transport of Contaminants in the Subsurface
Introduction
Subsurface transport of contaminants often is controlled by complex interactions between physical, chemical, and biological processes. The advection-dispersion equation used to quantitatively describe and predict contaminant movement in the subsurface also must contain reaction terms added to the basic equation to account for chemical and biological processes important in controlling contaminant transport and fate (Johnson and others, 1989).
Chemical Reactions of Organic Compounds
Chemical reactions may transform one compound into another, change the state of the compound, or cause a compound to combine with other organic or inorganic chemicals (Johnson and others, 1989). For use in the advection-dispersion equation, these reactions represent changes in the distribution of mass within the specified volume through which the movement of the chemicals is modeled.
Chemical reactions in the subsurface often are characterized kinetically as equilibrium, zero, or first order, depending on how the rate is affected by the concentrations of the reactants. A zero-order reaction is one that proceeds at a rate independent of the concentration of the reactant(s). In a first-order process, the rate of the reactions is directly dependent on the concentration of one of the reactants. The use of zero or first-order rate expressions may oversimplify the description of a process, but higher order expressions, which maybe more realistic, are often difficult to measure and/or model in complex environmental systems. Also first-order reactions are easy to incorporate into transport models (Johnson and others, 1989).
Sorption. Sorption is probably the most important chemical process affecting the transport of organic contaminants in the subsurface environment. Sorption of non-polar organics is usually considered an equilibrium-partitioning process between the aqueous phase and the porous medium (Chiou and others, 1979). When solute concentrations are low (i.e., either 510-5 Molar, or less than half the solubility, whichever is lower), partitioning often is described using a linear Freundlich isotherm, where the sorbed concentration is a function of the aqueous concentration and the partition coefficient (Kp) (Karickhoff and others,1979; Karickhoff, 1984). Kp usually is measured in laboratory batch equilibrium tests, and the data are plotted as the concentration in the aqueous phase versus the amount sorbed onto the solid phase (Figure 3-12) (Chiou and others, 1979).
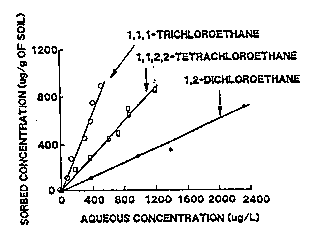
Figure 3-12. Batch Equilibrium Data for 1,1,1-TCA, 1,1,2,2,-TeCA and 1,2-DCA (adapted from Chiou and others, 1979, by Johnson and others, 1989)
Under conditions of linear equilibrium partitioning, the sorption process is represented in the advection dispersion equation as a “retardation factor,” R (Johnson and others, 1989). The retardation factor is dependent on the partition coefficient Kp, bulk density of aquifer materials, and porosity.
The primary mechanism of organic sorption is the formation of hydrophobic bonding between a contaminant and the natural organic matter associated with aquifers (Tanford, 1973; Karickhoff and others, 1979; Karickhoff,1984; Chiou and others,1985; MacKay and Powers, 1987). Therefore, the extent of sorption of a specific chemical can be estimated from the organic carbon content of the aquifer materials (foc) and a proportionality constant characteristic of the chemical (Koc), if the organic content is sufficiently high (i.e., fraction organic carbon content (foc) > 0.001) (Karickhoff and others,1979; Karickhoff,1984) . Koc values for many compounds are not known, so correlation equations relating Koc to more easily available chemical properties, such as solubility or octanol-water partition coefficients (Kenaga and Goring, 1980; Karickhoff, 1981; Schwarzenbach and Westall, 1981; Chiou and others, 1982,1983), have been developed. Within a compound class, Koc values derived from correlation expressions often can provide reasonable estimates of sorption. However, if correlations were developed covering a broad range of compounds, errors associated with the use of Koc estimates can be large (Johnson and others, 1989).
This method of estimation of sorption, using Koc and foc, values, is less expensive than the use of batch equilibrium tests. However, in soils with lower carbon content, sorption of neutral organic compounds onto the mineral phase can cause significant errors in the estimate of the partition coefficient (Chiou and others, 1985).
Hydrolysis. Hydrolysis, an important abiotic degradation process in ground water for certain classes of compounds, is the direct reaction of dissolved compounds with water molecules (Mabey and Mill, 1978). Hydrolysis of chlorinated compounds, which are often resistant to biodegradation (Siegrist and McCarty, 1987), forms an alcohol or alkene (Figure 3-13).
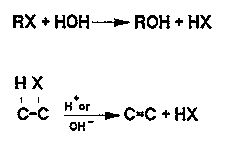
Figure 3-13. Schematic of Hydrolysis Reactions for Halogenated Organic Compounds (Johnson and others,1989).
Most information concerning rates hydrolysis is obtained from laboratory studies, since competing reaction and slow degradation rates make hydrolysis difficult to measure in the field. (Johnson and others,1989). Often data for hydrolysis are fitted as a first-order reaction, and a hydrolysis rate constant, K, is obtained. The rate constant multiplied by the concentration of the contaminant is added to the advection-dispersion equation to account for hydrolysis of the contaminant.
Cosolvation and Ionization. Cosolvation and ionization are processes that may decrease sorption and thereby increase transport velocity (Johnson and others,1989). The presence of cosolvents decreases entropic forces that favor sorption of hydrophobic organic contaminants by increasing interactions between the solute and the solvent (Nkedi-Kizza and others, 1985; Zachara and others, 1988). If biologically derived or anthropogenic solvent compounds are present at levels of 20 percent or more by volume, the solubility of hydrophobic organic contaminants can be increased by an order of magnitude or more (Nkedi-Kizza and others,1985). In Figure 3-14, decrease in sorption of anthracene in three soils, as described by the sorption coefficient Kp, is illustrated, with methanol as the cosolvent. Since cosolvent concentration must be large for solute velocity to be increased substantially, cosolvation is important primarily near sources of ground-water contamination.
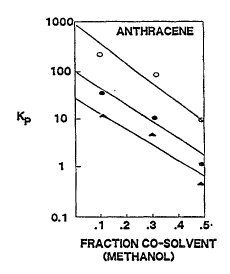
Figure 3-14. Effect of Methanol as a Cosolvent on Anthracene Soprtion for Three Soils (Adapted from Nkedi-Kizza and others, 1985, by Johnson and others, 1989).
In the process of ionization, acidic compounds, such as phenols or organic acids, can lose a proton in solution to form anions that, because of their charge, tend to be water-soluble (Zachara and others,1986). For example, the Koc of 2,4,5-trichlorophenol can decrease from 2,330 for the phenol, to almost zero for the phenolate (Figures 3-15 and 3-16) (Johnson and others, 1989). Acidic compounds tend to ionize more as the pH increases. However, for many compounds, such as the chlorophenols, substantial ionization can occurat neutral pH values.

Figure 3-15. Koc values for 2,4,5- trichlorophenolate (Johnson and others, 1989).
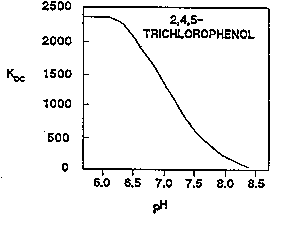
Figure 3-16. Koc versus pH for 2,4,5-trichlorophenol (Johnson and others, 1989).
Volatilization and Dissolution. Two important pathways for the movement of volatile organic compounds in the subsurface are volatilization into the unsaturated zone and dissolution into the ground water (Johnson and others, 1989). Contaminants in the aqueous and vapor phases are also more amenable to degradation.
The degree of volatilization of a contaminant is determined by: (1) the area of contact between the contaminated area and the unsaturated zone, which is affected by the nature of the medium (e.g., grain size, depth to water, water content) and the contaminant (e.g., surface tension and liquid density); (2) the vapor pressures of the contaminants; and (3) the rate at which the compound diffuses in the subsurface (Johnson and others, 1989).
The residual saturation remaining when immiscible liquids move downward through unsaturated porous media provides a large surface area for volatilization (Johnson and others, 1989). Vapor concentrations in the vicinity of the residual are often at saturation concentrations. Movement of vapor away from the residual saturation is usually controlled by molecular diffusion, which is affected by the tortuosity of the path through which the vapors move. Tortuosity also is affected by the air-filled porosity of the medium, so diffusion is reduced in porous media with a high water content.
Diffusion also is reduced by the partitioning of the vapors out of the gas phase and into the solid or aqueous phases (Johnson and others, 1989). The retardation factor developed for partitioning between the aqueous and solid phases can be modified with a term to describe partitioning between the vapor and aqueous phases.
When immiscible fluids reach the capillary fringe, their further movement is determined by the density of the fluids relative to water (Scheigg, 1984; Schwille, 1988). The LNAPLs pool on top of the water table while the DNAPLs penetrate into the ground water. Floating pools of LNAPL can provide substantial surface area for volatilization, with diffusion controlling the mass transfer of organic contaminants into the vapor phase.
The transport and fate of DNAPLs that penetrate into the ground water is controlled by dissolution. Experiments have shown that saturation concentration values can be maintained even with high ground-water velocities (e.g.,1 m/day) through a zone of contamination (Anderson and others,1987). During remedial activities, such as pump-and-treat, ground-water velocities may be high, but the dissolution process should still be effective.
Chemical Reactions of Inorganic Compounds
In studies of organic contamination,the most important characteristic is the total concentration of a contaminant in a certain phase (e.g., in water versus aquifer solid materials). However, studies of inorganic contamination are often more difficult because inorganic materials can occur in many chemical forms, and knowledge of these forms (i.e, species) is required to predict their behavior in ground water (Morel, 1983; Sposito, 1986).
In ground water, an inorganic contaminant may occur as: (1) “free ions” (i.e., surrounded only by water molecules); (2) insoluble species; (3) metal/ligand complexes; (4) adsorbed species; (5) species held on a surface by ion exchange; or (6) species differing by oxidation state (e.g., manganese (II) and (IV) or chromium (III) and (Vl) (Johnson and others, 1989).
The total concentration of an inorganic compound may not provide sufficient information to describe the fate and behavior of that compound in ground water. Mobility, reactivity, biological availability, and toxicity of metals and other inorganic compounds depend upon their speciation (Johnson and others, 1989). The primary reactions affecting the speciation of inorganic compounds are solubility and dissolution, complexation reactions, adsorption and surface chemistry, ion exchange, and redox chemistry.
Solubility, Dissolution, and Precipitation. Dissolution and weathering of minerals determine the natural composition of ground water (Johnson and others, 1989). Dissolution is the dissolving of all components within a mineral, while weathering is a partial dissolution process in which certain elements leach out of a mineral, leaving others behind.
Mineral dissolution is the source of most inorganic ions in ground water. In principle a mineral can dissolve up to the limits of its solubility, but in many cases, reactions occur at such a slow rate that true equilibrium is never attained (Morgan, 1967).
The contribution of ions from one mineral may affect the solubility of other minerals containing the same ion (i.e., the “common ion effect’). Computer programs such as MINTEQ (Felmy and others, 1984), MINEQL (Westall and others, 1976), and WATEQ2 (Ball and others, 1980) maybe used to predict the equilibrium distribution of chemical species in ground water and indicate if the water is undersaturated, supersaturated, or at equilibrium with various mineral phases. Some of these programs also may be used to predict the ionic composition of ground water in equilibrium with assumed mineral phases (Jennings and others, 1982).
The weathering of silicate minerals contributes cations, such as calcium, magnesium, sodium, potassium, and silica, to water and forms secondary weathering products such as kaolinite and montmorillonite clays (Johnson and others, 1989). This weathering increases the alkalinity of ground water to a level greater than its rainwater origins.
Weathering and dissolution also can be a source of contaminants. Leachates from mine tailings can yield arsenate, toxic metals, and strong mineral acids (Hem, 1970), while leachates from fly-ash piles can contribute selenium, arsenate, lithium, and toxic metals (Stumm and Morgan, 1981; Honeyman and others, 1982; Murarka and Macintosh, 1987).
The opposite of dissolution reactions is precipitation of minerals or contaminants from an aqueous solution (Johnson and others, 1989). During precipitation, the least-soluble mineral at a given pH level is removed from solution. An element is removed by precipitation when its solution concentration saturates the solubility of one of its solid compounds. If the solution concentration later drops below the solubility limit, the solid will begin to dissolve until the solubility level is attained again. Contaminants may initially precipitate, then slowly dissolve later after a remedial effort has reduced the solution concentration; thus complete remediation of the aquifer may require years.
A contaminant initially may be soluble but later precipitate after mixing with other waters or after contact with other minerals (Drever,1982; Williams, 1985; Palmer, 1989). For example, pumping water from an aquifer may mobilize lead until it converges and mixes with waters high in carbonates from a different formation and precipitates as a lead carbonate solid.
Compiexation Reactions. In complexation reactions, a metal ion reacts with an anion that functions as a ligand (Johnson and others, 1989). The metal and the ligand bind together to form a new soluble species called a complex. Transition metals form the strongest complexes (Stumm and Morgan, 1981); alkaline earth metals form only weak complexes, while alkali metals do not form complexes (Dempsey and O’Melia, 1983). The approximate order of complexing strength of metals is:
Fe(Ill)> Hg> Cu> Pb> Ni> Zn> Cd> Fe(¡¡» Mn> Ca> Mg
Common inorganic ligands that bind with metals include: OH-, CI-, SO4=, CO3=, S=, F–, NH3, PO4, CN–, and polyphosphates. Their binding strength depends primarily on the metal ion with which they are complexing (Johnson and others, 1989). Inorganic ligands are usually in excess compared to the “trace” metals with which they bind, and, therefore, they affect the fate of the metals in the environmental system, rather than vice versa (Morel, 1983).
Organic ligands generally form stronger complexes with metals than inorganic ligands (Johnson and others, 1989). Organic ligands include: (1) synthetic compounds from wastes, such as amines, pyridines, phenols, and other organic bases and weak acids; and (2) natural organic materials, primarily humic materials (Schnitzer, 1969; Hayes and Swift, 1978; Stevenson, 1982, 1985; Johnson and others, 1989). Humic materials are complex structures, and their complexation behavior is difficult to predict (Perdue and Lytle, 1983; Sposito,1984; Perdue, 1985; Dzombak and others, 1986; Fish and others, 1986). Generally, humic materials are found in significant concentrations only in shallow aquifers. In these aquifers, however, they may be the primary influence on the behavior of metals (Thurman, 1985).
Equilibrium among reactants and complexes for a given reaction is predicted by an equilibrium (or “stability”) constant, K, which defines a mass-law relationship among the species (Johnson and others, 1989). For given total ion concentrations (measured analytically), stability constants can be used to predict the concentration of all possible species (Martell and Smith, 1974, 1977; Smith and Martell, 1975).
Because complexes decrease the amount of free ions in solution, less metal may sorb onto aquifer solid materials or participate in precipitation reactions (Johnson and others, 1989). The metal is more soluble because it is primarily bound up in the soluble complex. Research has demonstrated that a metal undergoing complexation may be less toxic to aquifer microorganisms (Reuter and others, 1979).
Sorption and Surface Chemistry. Surface sorption, in many cases, is the most important process affecting toxic metal transport in the subsurface (Johnson and others, 1989). Changes in metal concentration, as well as pH, can have a significant effect on the extent of sorption (Figure 3-17).
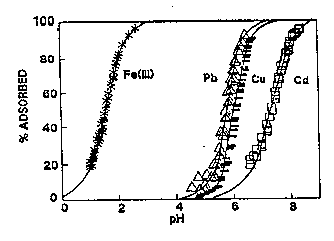
Figure 3-17. Adsorption of Metal Ions on Amorphous Silica as a Function of pH (adapted from Schindler and others, 1976, by Johnson and others, 1989).
Approaches to predicting behavior of metal ions based on sorption processes include using isotherms (indicating that data were collected at a fixed temperature) to graphically and mathematically represent sorption data (Johnson and others, 1989). Two types of isotherms are commonly used: the Freundlich isotherm and the Langmuir isotherm (Figure 3-18). The Freundlich isotherm is empirical, and sorbed (S) and aqueous (C) concentration data are fitted by adjusting two parameters (K and a). The Langmuir isotherm is based on the theory of surface complexation, using a parameter corresponding to the maximum amount that can be sorbed and the partition coefficient, K (Morel, 1983).
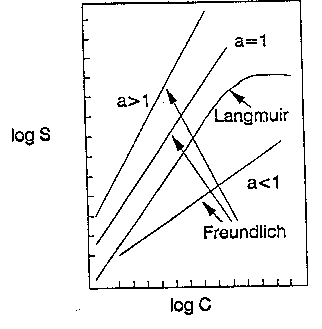
Figure 3-18. Schematic Representation of Freundlich and Langmuir Isotherm Shapes for Batch Equilibrium Tests (Johnson and others, 1989).
Another method to describe sorption is to use surface complexation models that represent sorption as ions binding to specific chemical functional groups on a reactive surface (Johnson and others,1989). All surface sites may be identical or may be grouped into different classes of sites (Benjamin and Leckie, 1981). Each type of site has a set of specific sorbing constants, one for each sorbing compound. Electrostatic forces at the surface also contribute to the overall sorption constant (Davis and others, 1978). Binding of ions to the surface is calculated from constants using mass-law equations similar to those used to calculate complex formation (Schindler and others, 1976; Stumm and others, 1976; Dzombak and Morel, 1986). However, the parameters used in surface complexation models are data-fitting parameters, which fit a specified set of data to a particular model, but have no thermodynamic meaning and no generality beyond the calibrating data set (Westall and others, 1980).
Ion-Exchange Reactions. Ion-exchange reactions are similar to sorption. However, sorption is coordination bonding of metals (or anions) to specific surface sites and is considered to be two-dimensional, while an ion-exchanger is a three-dimensional, porous matrix containing fixed charges (Helfferich, 1962; Johnson and others, 1989). Ions are held by electrostatic forces rather than by coordination bonding. Ion-exchange “selectivity coefficients” are empirical and vary with the amount of ion present (Reichenburg, 1966). Ion exchange is used to describe the binding of alkali metals, alkaline earths, and some anions to clays and humic materials (Helfferich, 1962; Sposito, 1984). Knowledge of ion exchange is used to understand the behavior of major natural ions in aquifers and also is useful for understanding behavior of contaminant ions at low levels. In addition, ion exchange models are used to represent competition among metals for surface binding (Sposito, 1984).
Redox Chemistry. Reduction-oxidation (redox) reactions involve a change in the oxidation state of elements (Johnson and others, 1989). The amount of change is determined by the number of electrons transferred during the reaction (Stumm and Morgan, 1981). The oxidation status of an element can be important in determining the potential for transport of that element. For example, in slightly acidic to alkaline environments, Fe(III) precipitates as a highly sorptive phase (ferric hydroxide), while Fe(II) is soluble and does not retain other metals. The reduction of Fe(III) to Fe(II) releases not only Fe+2 to the water, but also other contaminants sorbed to the ferric hydroxide surfaces (Evans and others, 1983; Sholkovitz, 1985).
Chromium (Cr) (VI) is a toxic, relatively mobile anion, while Cr (III) is immobile, relatively insoluble, and strongly sorbs to surfaces. Selenate (Se) (VI) is mobile but less toxic, while selenite Se(IV) is more toxic but less mobile (Johnson and others, 1989).
The redox state of an aquifer is usually closely related to microbial activity and the type of substrates available to the microorganisms (Johnson and others, 1989). As organic contaminants are oxidized in an aquifer, oxygen is depleted and chemically reducing (anaerobic) conditions form. The redox reactions that occur depend on the dominant electron potential, which is defined by the primary redox-active species. The combination of Fe(II)/Fe(III) defines a narrow range of electron potentials, while (S) (sulfur)(+IV)/S(-II) defines a broader range. Pairs of chemical species are called redox couples.
After oxygen is depleted from ground water, the most easily reduced materials begin to react and, along with the reduced product, determine the dominant potential. After that material is reduced, the next most easily reduced material begins to react. These series of reactions continue, usually catalyzed by microorganisms. An aquifer may be described as “mildly reducing” or “strongly reducing,” depending on where it is in the chemical series (Stumm and Morgan, 1981).
The electron potential of water may be measured in volts, as Eh, or expressed by the “pe,” which is the negative logarithm of the electron activity in the water (Johnson and others, 1989). A set of redox reactions is often summarized on a pH-pe (or pH-Eh) diagram, which shows the predominant redox species at any specified pH and pe (or Eh). In this theoretical approach, only one redox couple should define the redox potential of the system at equilibrium. However, in an aquifer, many redox couples not in equilibrium can be observed simultaneously (Lindberg and Runnels, 1984). Therefore, redox behavior of chemicals in aquifers is difficult to predict. However, the redox status of an aquifer is important because of its effects on the mobility of elements and the potential effects on biodegradation of organic contaminants. Anaerobic (reducing) conditions are not favorable for hydrocarbon degradation, but reducing conditions favor dehalogenation of chlorinated and other halogenated compounds (Johnson and others, 1989).
Biological Processes Controlling the Transport of Contaminants In the Subsurface
Introduction
Historically, ground water was thought to be a safe water source because it was protected by a metabolically diverse “living filter” of microorganisms in the soil root zone that converted organic contaminants to innocuous end-products (Suflita,1989a). Aquifers were considered to be abiotic environments, based on studies that showed that microbial numbers decreased with soil depth (Waksman, 1916) and that indicated that most microorganisms were attached to soil particles (Balkwill and others, 1977). In addition, by estimating the time required for surface water to vertically penetrate subsurface formations, researchers felt that microorganisms travelling with water would utilize available nutrients and rapidly die off. Therefore, since aquifers were considered to be sterile, they could not be biologically remediated if contaminated with organic contaminants. However, microscopic, cultivation, metabolic, and biochemical investigations, using aseptically obtained aquifer materials, have shown that there are high numbers of metabolically diverse procaryotic and eucaryotic organisms present in the terrestrial subsurface environment (Suflita, 1989a).
Evidence of Subsurface Microorganisms
Microbiological investigations have detected high numbers of microorganisms (up to 50 x 106 total cells/mL) in both contaminated and uncontaminated aquifers at various depths and geological composition (Suflita, 1989a). Even deep geological formations may be suitable habitats for microorganisms (Kuznetsov and others, 1963; Updegraff, 1982). The microorganisms that have been detected in the subsurface are small, capable of response to addition of nutrients, and are primarily attached to solid surfaces. Eucaryotic organisms are present in the subsurface but are few in numbers and are probably of minor significance, existing as inert resting structures (Suflita, 1989a).
Suitable sampling technology was developed to demonstrate the existence of subsurface microorganisms (Suflita, 1989a). Samples must not be contaminated with nonindigenous microorganisms originating from drilling machinery, surface soil layers, drilling muds, and water used to make up drilling muds. Since most subsurface microorganisms are associated with aquifer solid materials, current sampling efforts use core recovery and dissection to remove microbiologically contaminated portions of the cores (McNabb and Mallard, 1984). This dissection is performed in the field, to prevent nonindigenous organisms from penetrating to the inner portions of the core, or in the laboratory if it is nearby. The outer few centimeters and the top and bottom portions of the aquifer cores are removed because of possible contamination by nonindigenous bacteria, and the center portions of the cores are used for microbiologicalanalysis. An alcohol-sterilized paring device is used in the dissection process. The paring device has an inner diameter that is smaller than the diameter of the core itself. As the aquifer material is extruded out of the sampling core barrel and over the paring device, the potentially contaminated material is stripped away. For anaerobic aquifers, this field paring dissection is performed inside plastic anaerobic glove bags while the latter is purged with nitrogen to minimize exposure of the microorganisms to oxygen (Beeman and Suflita,1987). Samples obtained by this technique are considered to be aseptically acquired and are suitable for microbiological analyses.
Evidence of Activity of Subsurface Microorganisms
Although direct and conclusive evidence had been obtained about the existence of microorganisms in the subsurface, questions remained about their significance in ground water. Such questions included: (1) whether or not the indigenous microorganisms were metabolically active, (2) what was the diversity of the metabolic activities, (3) what factors served to limit and/or stimulate the growth and metabolism of these organisms, and (4) could the inherent metabolic versatility of aquifer microorganisms be utilized to remediate contaminated aquifers (Suflita, 1989a).
Microbial subsurface activity was studied, and the following metabolic processes were identified in the subsurface environment: (1) biodegradation of organic pollutants, including petroleum hydrocarbons, alkylpyridines, creosote chemicals, coal gasification products, sewage effluent, halogenated organic compounds, nitriloacetate (NTA), and pesticides; (2) nitrification; (3) denitrification; (4) sulfur oxidation and reduction; (5) iron oxidation and reduction; (6) manganese oxidation; and (7) methanogenesis (Suflita, 1989a). These metabolic processes include aerobic and anaerobic carbon transformations, many of which are important in aquifer contaminant biodegradation. The other processes are those required for the cycling of nitrogen, sulfur, iron, and manganese in microbial communities.
Biodegradation may refer to complete mineralization of organic contaminants (i.e., the parent compounds), to carbon dioxide, water, inorganic compounds, and cell protein (Sims and others, 1990). The ultimate products of aerobic metabolism are carbon dioxide and water, while under anaerobic conditions, metabolic activities also result in the formation of incompletely oxidized simple organic substances such as organic acids and other products such as methane or hydrogen gas.
Since contaminant biodegradation in the natural environment is frequently a stepwise process involving many enzymes and many species of organisms, a contaminant may not be completely degraded. Instead, it may be transformed to intermediate product(s) that may be less, equally, or more hazardous than the parent compound, and more or less mobile in the environment (Sims and others, 1990). The loss of a chemical, therefore, may or may not be a desirable consequence of the biodegradation process if biodegradation results in the production of undesirable metabolites with their own environmental impact and persistence characteristics (Suflita, 1989b). For example, the reductive removal of tetrachloroethylene (TeCE) under anaerobic conditions results in a series of dehalogenated intermediates. TeCE’s halogens are removed and replaced by protons in a series of sequential steps. However, the rate of reductive dehalogenation decreases as fewer and fewer halogens remain. Consequently, highly toxic vinyl chloride accumulates and, from a regulatory standpoint, causes greater concern than the parent contaminant. Bioremedial technologies should be selected with knowledge of metabolic processes of the specific contaminants at the site.
Biodegradation of most organic compounds in aquifer systems may be evaluated by monitoring their disappearance from the aquifer through time. Disappearance, or rate of degradation, is often expressed as a function of the concentration of one or more of the contaminants being degraded (Sims and others, 1990). Biodegradation in natural systems often can be modeled as a first-order chemical reaction (Johnson and others, 1989). Both laboratory and field data suggest that this is true when none of the reactants are in limited supply. A useful term to describe reaction kinetics is the half-life, t1/2, which is the time required to transform 50 percent of the initial constituent.
As decomposable organic matter enters an oxygenated aquifer (Figure 3-19), microbial metabolism will likely begin to degrade the contaminating substrate; i.e., the indigenous microorganisms utilize the contaminant as an electron donor for heterotrophic microbial respiration (Suflita,1989a). The aquifer microorganisms use oxygen as a co-substrate and as an electron acceptorto support their respiration. This oxygen demand may deplete oxygen and establish anaerobic conditions. When oxygen becomes limiting, aerobic respiration slows, and other microorganisms become active and continue to degrade the organic contaminants. Under conditions of anoxia, anaerobic bacteria use organic chemicals or certain inorganic anions as alternate electron acceptors.
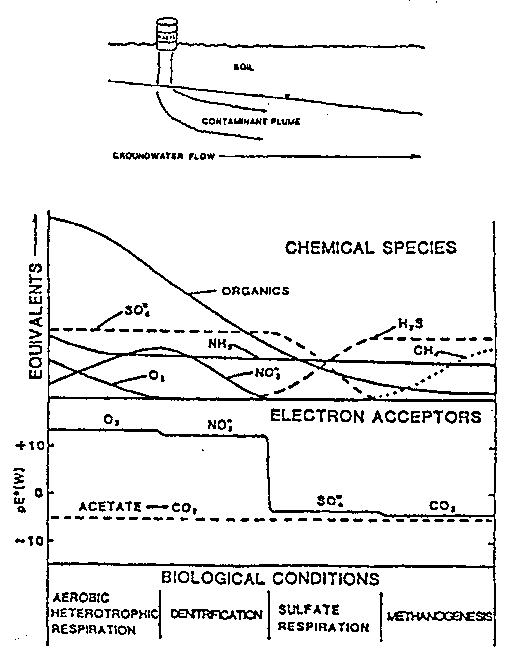
Figure 3-19. Microbially Mediated Changes in Chemical Species, Redox Conditions, and Spatial Regions Favoring Different Types of Metabolic Processes Along the Flow Path of a Contaminant Plume (adapted from Bouwer and McCarty, 1984, by Suflita, 1989a).
Nitrate present in ground water is not rapidly depleted until oxygen is utilized. Organic matter is still metabolized, but, instead of oxygen, nitrate becomes the terminal electron acceptor during denitrification. Sulfate becomes a terminal electron acceptor when nitrate is limiting. When this occurs, hydrogen sulfide, an odorous gas, can often be detected in the ground water as a metabolic end-product. When very highly reducing conditions are present in an aquifer, carbon dioxide becomes an electron acceptor and methane is formed. Sometimes a spatial separation of dominant metabolic processes can occur in an aquifer, depending on the availability of electron acceptors, the presence of suitable microorganisms, and the energy benefit of the metabolic process to the specific microbial communities. As organic matter is transported in a contaminant plume, a series of redox zones can be established that range from highly oxidized to highly reduced conditions. The biodegradation potential and the expected rates of metabolism will be different in each zone (Suflita,1989a). For many contaminants, aerobic decomposition is relatively fast, especially compared to methanogenic conditions. However, some contaminants, such as certain halogenated aliphatic compounds and 2,4,5-T, degrade faster when anaerobic conditions exist (Bouwer and others, 1981; Bouwer and McCarty, 1984; Gibson and Suflita, 1986).
Environmental Factors Aftecting Biodegradation
Microorganisms need a suitable physical and chemical environment to grow and actively metabolize organic contaminants, (Suflita,1989a). Extremes of temperature, pH, salinity, osmotic or hydrostatic pressures, radiation, free water limitations, contaminant concentration, and/or the presence of toxic metals or other toxicant materials can limit the rate of microbial growth and/or substrate utilization. Often, two or more environmental factors interact to limit microbial decomposition processes. Selected critical environmental factors are presented in Table 3-1.
| Environmental Factor | Optimum Levels |
| Avalilable soil water | 25-85% of water holding capacity; -0.01 MPs |
| Oxygen | Aerobic metabolism: Greater than 0.2 mg/l dissolved oxygen, minimum air-filled pore space of 10% by volume; Anaerobic metabolism: O2 concentrations less than 1% by volume |
| Redox potential | Aerobes & facultative anaerobes:greater than 50 millivolts; Anaerobes: less than 50 millivolts |
| pH | pH values of 5.5 – 8.5 |
| Nutrients | Sufficient nitrogen, phosphorus, and other nutrients so as to not limit microbial growth (Suggested C:N:P ratio of 120:10:1) |
| Temperature | 15 – 45° C (Mesophiles) |
Table 3-1. Critical Environmental Factors for Microbial Activity (Sims and others, 1984; Huddleston and others, 1986; Paul and Clark, 1989).
Limitations in the ability to alter environmental factors in the subsurface environment are important in selecting and implementing aquifer bioremedial technologies (Suflita,1989a). For example, the temperature of aquifers probably cannot be significantly altered to stimulate in situ microbial growth and metabolism, but temperatures could be changed in a surface biological treatment reactor.
Physiological Factors Affecting Biodegradation
In addition to environmental conditions, microbial physiological factors also influence organic contaminant biodegradation (Suflita, 1989a). The supply of carbon and energy contained in organic contaminants must be sufficient for heterotrophic microbial growth. Too high a substrate concentration can limit microbial metabolism due to the toxicity of the substrate to microorganisms. If concentrations are too low, microbial response may be inhibited, or the substrates may not be suitable for growth. Growth and energy sources do not have to be supplied by the same carbon substrate. Growth and metabolism of microorganisms can be stimulated by providing a non-toxic primary carbon substrate so that the rate and extent of contaminant degradation can be increased (McCarty and others,1981; McCarty, 1985; McCarty and others, 1984).
A contaminant also will be poorly metabolized if it is unable to enter microbial cells and gain access to intracellular metabolic enzymes, which may occur with larger molecular weight compounds (Suflita, 1989a). A substrate also will persist if it fails to de-repress the enzymes required for its degradation. Appropriate enzymes sometimes can be induced by an alternate chemical compound. Sometimes initial biochemical reactions result in metabolites that tend to inhibit degradation of the parent molecule.
The absence of other necessary microorganisms can limit contaminant degradation, since often several microbial groups are required for complete degradation (Suflita, 1989a). Microbial consortia are especially important in anaerobic mineralization of contaminants (Mclnerney and Bryant, 1981); if any individual members of a consortium are absent, biodegradation of the parent material effectively ceases.
Chemical Factors Affecting Biodegradation
One of the most important factors affecting contaminant biodegradation in aquifers is the structure of the contaminant, which determines its physical state (i.e., soluble, sorbed) and its tendency to biodegrade (Suflita, 1989a). Aquifer contaminants may contain chemical linkages that tend to favor or hinder microbial degradation. The number, type, and position of substituents on a contaminant molecule should be considered when evaluating its metabolic fate in an aquifer.
Usually the closer a contaminant structurally resembles a naturally occurring compound, the better the possibility that the contaminant will be able to enter a microbial cell, de-repress the synthesis of metabolic enzymes, and be converted by those enzymes to metabolic intermediates (Suflita, 1989a). Biodegradation is less likely (though not precluded) for those molecules having unusual structural features infrequently encountered in the natural environment. Therefore, xenobiotic compounds tend to persist in the natural environment because microorganisms have not evolved necessary metabolic pathways to degrade those compounds. However, microorganisms are nutritionally versatile, have the potential to grow rapidly, and possess only a single copy of DNA. Therefore, any genetic mutation or recombination is immediately expressed. If the alteration is of adaptive significance, new species of microorganisms can be formed and grow. Contaminated environments supply selection pressure for the evolution of organisms with new metabolic potential that can grow utilizing the contaminating substance.
Aquifer Bioremediation
If an aquifer contaminant is determined to be susceptible to biodegradation, the goal of bioremediation is to utilize the metabolic capabilities of the indigenous microorganisms to eliminate that contaminant (Suflita, 1989a). This practice generally does not include the inoculation of the aquifer with foreign bacteria.
Bioremedial technologies attempt to impose particular conditions in an aquifer to encourage microbial growth and the presence of desirable microorganisms. Bioremediation is based on knowledge of the chemical and physical needs of the microorganisms and the predominant metabolic pathways (Suflita,1989a). Most often, microbial activity is stimulated by supplying nutrients necessary for microbial growth. Bioremediation can take place either above ground or in situ. In situ systems are especially appropriate for contaminants that sorb to aquifer materials, since many decades of pumping may be required to reduce the contaminants to sufficiently low levels.
Successful implementation of aquifer bioremediation depends on determining site-specific hydrogeological variables, such as type and composition of an aquifer, permeability, thickness, interconnection to other aquifers, location of discharge areas, magnitude of water table fluctuations, and ground-water flow rates (Suflita, 1989b). Generally, bioremediation is utilized in more permeable aquifer systems where movement of ground water can be more successfully controlled.
Removal of free product also is important for the success of bioremediation. Many substances that serve as suitable nutrients for microbial growth when present at low concentrations are inhibitory at high concentrations (Suflita, 1989b).
Modeling Transport and Fate of Contaminants In an Aquifer
Introduction
Models are simplified representations of real-world processes and events, and their creation and use require many judgments based on observation of simulations of specific natural processes. Models may be used to simulate the response of specific problems to a variety of possible solutions (Keely, 1989b).
Physical models, including sand-filled tanks used to simulate aquifers and laboratory columns used to study contaminant flow through aquifer materials, often are used to obtain information on contaminant movement (Keely, 1989b). Analog models also are physically based, but are only similar to actual processes. An example is the electric analog model, where capacitors and resistors are used to replicate the effects of the rate of water release from storage in aquifers. The main disadvantage of physical models is the time and effort required to generate a meaningful amount of data.
Mathematical models are non-physical and rely on quantification of relationships between specific parameters and variables to simulate the effects of natural processes (Keely, 1989b, Weaver and others, 1989). Because mathematical models are abstract, they often do not provide an intuitive knowledge of real-world situations. However, mathematical models can provide insights into the functional dependencies between causes and effects in an actual aquifer. Large amounts of data can be generated quickly, and experimental modifications made easily, making possible for many situations to be studied in detail for a given problem.
Use and Categories of Mathematical Models
The application of mathematical models is subject to error in real-world situations when appropriate field determinations of natural process parameters are lacking. This source of error is not addressed adequately by sensitivity analyses or by the application of stochastic techniques for estimating uncertainty. The high degree of hydrogeological, chemical, and microbiological complexity typically present in field situations requires the use of site-specific characterization of the influences of various natural processes by detailed field and laboratory investigations (Keely, 1989b).
Mathematical models have been categorized by their technical bases and capabilities as: (1) parameter identification models; (2) prediction models; (3) resource management models; and (4) data manipulation codes. (Bachmat and others, 1978; van der Heidje and others, 1985).
Parameter identification models are used to estimate aquifer coefficients that determine fluid flow and contaminant transport characteristics (e.g., annual recharge, coefficients of permeability and storage, and dispersivity (Shelton, 1982; Guven and others, 1984; Puri, 1984; Khan, 1986a, b; Strecker and Chu, 1986)). Prediction models are the most numerous type because they are the primary tools used for testing hypotheses (Mercer and Faust, 1981; Anderson and others, 1984; Krabbenhoft and Anderson, 1986).
Resource management models are combinations of predictive models, constraining functions (e.g., total pumpage allowed), and optimization routines for objective functions (e.g., scheduling wellfield operations for minimum cost or minimum drawdown/pumping lift). Few of these types of models are developed well enough or supported to the degree that they are useful (van der Heidje,1984a and b; van derHeidje and others, 1985).
Data manipulations codes are used to simplify data entry to other kinds of models and facilitate the productions of graphic displays of model outputs (van der Heidje and Srinivasan, 1983; Srinivasan, 1984; Moses and Herman, 1986).
Quality Control Measures
Quality control measures are required to assess the soundness and utility of a mathematical model and to evaluate its application to a specific problem. Huyakorn et al. (1984) and Keely (1989b) have suggested the following quality control measures:
- Validation of the model’s mathematical basis by comparing its output with known analytical solutions to specific problems.
- Verification of the model’s application to various problem categories by successful simulation of observed field data.
- Benchmarking the problem-solving efficiency of a model by comparison with the performance of other models.
- Critical review of the problem conceptualization to ensure that the modeling considers all physical, chemical, and biological processes that may affect the problem.
- Evaluation of the specifics of the model’s application, e.g., appropriateness of the boundary conditions, grid design, time steps.
- Appraisal of the match between the mathematical sophistication of the model and the temporal and spatial resolution of the data.
Summary
Transport and fate assessments require interdisciplinary analyses and interpretations because processes are interdependent (Keely 1989a). Each transport process should be studied from interdisciplinary viewpoints, and interactions among processes identified and understood. In addition to a sound conceptual understanding of transport processes, the integration of information on geologic, hydrologic, chemical, and biological processes into an effective contaminant transport evaluation requires data that are accurate, precise, and appropriate at the intended problem scale and that attempt to account for spatial and temporal variations.
References
Anderson, M. A., J. F. Pankow, and R. L. Johnson, 1987, The dissolution of residual dense non-aqueous phase liquid (DNAPL) from a saturated porous medium: jn Proceedings, Petroleum Hydrocarbons and Organic Chemicals in Ground Water. Nat. Water Well Assn and the American Petrol. Institute, Houston, TX, November, pp. 409-428.
Anderson, P. F., C. R. Faust, and J. W. Mercer, 1984, Analysis of conceptual designs for remedial measures at Lipari Landfill, New Jersey: Ground Water, v. 22, pp. 176-190.
Bachmat, Y., B. Andrews, D. Holtz, and S. Sebastian, 1978, Utilization of numerical groundwater models for Water Resource Management: EPA-600/8-78-012.
Balkwill, D. L., T. E. Rucinski, and L. E. Casida, Jr.,1977, Release of microorganism from soil with respect to transmission electron microscopy viewing and plate counts: Antonie Van Leeuwenhoek ed., Journal of Microbiology and Serology, v. 43, pp. 73-87.
Ball, J. W., D. K. Nordstrom, and E. A. Jenne, 1980, Additional and revised thermochemical data and computer code for WATEQ2: A Computerized Model for Trace and Major Element Speciation and Mineral Equilibria of Natural Waters. Water Resources Investigations U. S. Geological Survey, no. 78-116.
Bear, J., 1969, Hydrodynamic dispersion: ice, Flow Through Porous Media, R.J.M. DeWiest, Editor: Academic Press, New York, pp. 109-199.
Bear, J., 1979, Hydraulics of groundwater: McGrawHill, New York.
Beeman, R. E. and J. M. Suflita, 1987, Microbial ecology of a shallow unconfined ground-water aquifer polluted by municipal landfill leachate: Microbial Ecology, v. 14, pp. 39-54.
Benjamin, M. M. and J. O. Leckie, 1981, Multiple site adsorption of Cd, Cu, Zn, and Pb on amorphous iron oxyhyroxides: Journal of Colloid and Interface Science, v. 79, no. 2, pp. 209-221.
Bouwer, E. J. and P. L. McCarty, 1984, Modeling of trace organics biotransformation in the subsurface: Ground Water, v. 22, pp. 433-440.
Bouwer, E. J., B. E. Rittmann, and P. L. McCarty, 1981, Anaerobic degradation of halogenated 1-and2-carbon organic compounds: Environmental Science and Technology, v.15, pp. 596-599.
Chiou, C. T., D. W. Schmedding, and M. Manes, 1982, Partitioning of organic compounds on octanol-water systems: Environmental Science and Technology, v. 16, pp. 4-10.
Chiou, C. T., L. J. Peters, and V. H. Freed, 1979, A physical concept of soil-water equilibria for nonionic compounds: Science, v. 206, pp. 831-832.
Chiou, C. T., P. E. Porter, and D. W. Schmedding,1983, Partition equilibria of nonionic organic compounds between soils organic matter and water: Environmental Science and Technology, v. 17, pp. 227-231.
Chiou, C. T., T. D. Shoup, and P. E. Porter, 1985, Mechanistic roles of soil humus and minerals in the sorption of nonionic organic compounds from aqueous and organic solutions: Organic Geochemistry, v. 8, pp. 9-14.
Chouke, R. L., P. van Meurs, and C. van der Poel,1959, The instability of slow, immiscible, viscous liquid-liquid displacements in permeable media transactions: American Institute of Mining Engineers, v. 216, pp.188-194.
Davis, J. A., R. O. James, and J. O. Leckie, 1978, Surface ionization and complexation at the oxide/water interface: I. computation of electrical double layer properties in simple electrolytes: Journal of Colloid and Interface Science, v. 63. no. 3, pp. 480-499.
Dempsey, B. A. and C. R. O’Melia, 1983, Proton and calcium complexation of four fulvic acid Factions: ID, Aquatic and Terrestrial Humic Materials, R. F. Christman and E. T. Gjessing, Editors. Ann Arbor Science, Ann Arbor, M I.
Drever, J.1.,1982, The geochemistry of natural waters: Prentice-Hall, Englewood Cliffs, NJ.
Dzombak, D. A. and F. M. M. Morel, 1986, Sorption of cadmiumon hydrous ferric oxide at high sorbate/sorbent ratios: equilibrium, kinetics, and modelling: Journal of Colloid and Interface Science, v. 112, no. 2, pp. 588-598.
Dzombak, D. A., W. Fish, and F. M. M. Morel, 1986, Metal-humate interactions. 1. discrete ligand and continuous distribution models: Environmental Science and Technology, v. 20, pp. 669-675.
Evans, D. W., J. J. Alberts, and R. A. Clark, 1983, Reversible ion-exchange of cesium-137 leading to mobilization from reservoir sediments: Geochemica et Cosmochimica Acta, v. 47, no. 11, pp. 1041-1049.
Felmy, A. R., D. C. Girvin, and E. A. Jenne. 1984. MINTED: A computer program for calculating aqueous geochemical equilibria: EPA/600/3-84-032, U. S. Environmental Protection Agency, Environmental Research Laboratory, Athens, GA.
Feenstra, S. and J. A. Cherry, 1987, Dense organic solvents in ground water: an introduction: In Dense Chlorinated Solvents in Ground Water, Progress Report No. 083985, Institute for Ground Water Research, University of Waterloo, Ontario.
Fish, W., D. A. Dzombak, and F. M. M. Morel, 1986, Metal-humate interactions. 2. application and comparison of models: Environmental Science and Technology, v. 20, pp. 676-683.
Frind, E. O. and G. E. Hokkanen, 1987, Simulation of the borden plume using the alternating direction galerkin technique: Water Resources Research, v. 23, no. 5, pp. 918-930.
Gibson, S. A. and J. M. Suflita, 1986, Extrapolation of biodegradative results to groundwater aquifers: reductive dehalogenation of aromatic compounds: Applied and Environmental Microbiology, v. 52:681-688.
Gillham, R. W., and J. A. Cherry, 1982, Contaminant migration in saturated unconsolidated geologic deposits: in Recent Trends in Hydrogeology, T. N. Narasimhan, Editor. Geological Society of America, Paper 189, pp. 31-62, Boulder, CO.
Guven, O., F. J. Molz, and J. G. Melville, 1984, An analysis of dispersion in a stratified aquifer: Water Resources Research, v. 20, pp. 1337-1354.
Hayes, M. H. B. and R. S. Swift, 1978, The chemistry of soil organic colloids: In The Chemistry of Soil Constituents, D. J. Greenland and M. H. B. Hays, Editors. Wiley Interscience, New York, NY.
Helfferich, F., 1962, Ion exchange: McGraw-Hill, New York, NY.
Hem, J. D.,1970, Study and interpretation of the chemical characteristics of natural water: Water Supply Paper 1473, U. S. Geological Survey, Reston, VA.
Homsy. G. M.,1987, Viscous fingering in porous media: Annual Review of Fluid Mechanics, v. 19, pp. 271-311.
Honeyman, B. D., K. F. Hayes, and J. O. Leckie, 1982, Aqueous chemistry of As, B, Cr, Se, and V with particular reference to fly-Ash transport water: Project Report EPRI-910-1, Electric Power Research Institute, Palo Alto, CA.
Huddleston, R. L., C. A. Bleckmann, and J. R. Wolfe, 1986, Land treatment biological degradation processes: In Land Treatment: A Waste Management Alternative, R. C. Loehr and J. F. Malina, Jr., Editors. Water Resources Symposium, Center for Research in Water Resources, The University of Texas at Austin, Austin, TX, No. 13, pp. 41-61.
Huyakorn, P. S., and others, 1984, Testing and validation of models for simulating solute transport in ground water: development and testing of benchmark techniques. IGWMC: Report No. GWMI 84-13. International Ground Water Modeling Center, Holcolm Research Institute, Butler University, Indianapolis, IN.
Johnson, R. L., C. D. Palmer, and W. Fish. 1989. Subsurface chemical processes: In Transport and Fate of Contaminants in the Subsurface. U.S. Environmental Protection Agency, Centerfor Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/ 625/4-89/019.
Karickhoff, S. W., 1981, Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils: Chemosphere, v. 10, pp. 833-846.
Karickhoff, S. W., 1984, Organic pollutant sorption on aquatic systems: Journal of Hydraulic Engineering, v. 10, pp. 833-846.
Karickhoff, S. W., D. S. Brown, and T. A. Scott, 1979, sorption of hydrophobic pollutants on natural sediments: Water Research, v. 13, pp. 241-248.
Kenaga, E. E. and C. A. I. Goring, 1980, Relationship between water solubility, soil sorption, octanol-water partitioning, and concentrations of chemicals in Biota: jg Aquatic Toxicology, Third Conference, J. G. Eaton, P. R. Parrish, and A. C. Hendricks, Editors. ASTM Special Publication 707. American Society for Testing Materials, Philadelphia, PA., pp. 78-115.
Keely, J. F., 1989a, Introduction: in Transport and Fate of Contaminants in the Subsurface. U.S.Environmental Protection Agency, Centerfor Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/ 625/4-89/019.
Keely, J. F., 1989b, Modeling subsurface contaminant transport and fate: in Transport and Fate of Contaminants in the Subsurface. U.S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/625/4-89/019.
Khan, 1. A., 1986a, Inverse problem in ground water: model development: Ground Water, v. 24, pp. 32-38.
Khan, I. A., 1986b, Inverse problem in ground water: model application: Ground Water, v. 24, pp. 39-48.
Kimmel, G. E. and O. C. Braids, 1980, Leachate plumes in groundwater from Babylon and Islip landfills, Long Island, New York: Professional Paper 1085, U. S. Geological Survey, Reston, VA.
Krabbenhoft, D. P. and M. P. Anderson, 1986, Use of a numerical ground-water flow model for hypothesis testing: Ground Water, v. 24, pp. 49-55.
Kueper, B. H. and E. O. Frind, 1988, An overview of immiscible fingering in porous media: Journal of Contaminant Hydrology, v. 2, pp. 95-110.
Kuznetsov, S. I., N. V. Ivanov, and N. N. Lyalikova, 1963, The distribution of bacteria in groundwaters and sedimentary rocks: in Introduction to Geological Microbiology, C. Oppenheimer, Editor. McGraw-Hill Book Co., New York, NY.
Lindberg, R. D. and D. D. Runnels, 1984, Ground water redox reactions: An analysis of equilibrium state applied to Eh measurements and geochemical modeling: Science v. 225, pp. 925-927.
Mabey, W. R. and T. Mill, 1978, Critical review of hydrolysis of organic compounds in water under environmental conditions: Journal of Physical and Chemical Reference Data, v. 7, pp. 383-415.
MacFarlane D. S., J. A. Cherry, R. W. Gillham, and E. A. Sudicky, 1983, Migration of contaminants in groundwater at a landfill: A case study; 1. groundwater flow and plume delineation: Journal of Hydrology, v. 63, pp. 1-29.
MacKay, D. and B. Powers, 1987, Sorption of hydrophobic chemicals from water: A hypothesis for the mechanism of the particle concentration effect: Chemosphere, v. 16, pp. 745-757.
Martell, A. E. and Smith, R. M., 1974, Critical stability constants: Amino Acids, v.1, Plenu m Press, New York, NY.
Martell, A. E. and Smith, R. M., 1977., Critical stability constants: Other Organic Ligands, v. 3, Plenum Press, New York, NY.
McCarty, P. L., 1985, Application of biological transformations in groundwater: in Proceedings, Second International Conference on Ground-Water Quality Research, N. N. Durham and A. E. Redelfs, Editors. National Center for Groundwater Research, Stillwater, OK.
McCarty, P. L., B. E. Rittmann, and E. J. Bouwer,1984, Microbiological processes affecting chemical transformations in ground water: in Groundwater Pollution Microbiology, G. Bitton and C. P. Gerba, Editors. John Wiley & Sons, New York, NY.
McCarty, P. L., M. Reinhard, and B. E. Rittmann, 1981, Trace organics in groundwater: Environmental Science and Technology, v. 15, pp. 47-51.
McDowell-Boyer, L. M., J. R. Hunt, and N. Sitar, 1986, Particle transport through porous media: Water Resources Research, v. 22, no. 13, pp. 1901-1921.
McInerney, M. J. and M. P. Bryant, 1981, Basic principles of bioconversion in anaerobic digestion and methanogenesis: in Biomass Conversion Processes for Energy and Fuels, S. S. Sofer and O. R. Zaborsky, Editors. Plenum Publishing Corporation, New York, NY, pp. 277-296.
McNabb, J. F. and G. E. Mallard, 1984, Microbiological sampling in the assessment of groundwater pollution: in Groundwater Pollution Microbiology, G. Bitton and C. P. Gerba, Editors, John Wiley & Sons, New York, NY, pp. 235-260.
Mercado, A., 1967, The spreading pattern of injected water in a permeability stratified aquifer: jn Proceedings of the Symposium on Artificial Recharge and Management of Aquifers, Haifa, Israel, May 19-26. Publication No. 72, International Association of Scientific Hydrology, Gentbruuge, Belgium, pp. 23-36.
Mercer, J. W. and C. R. Faust, 1981; Ground-water modeling: National WaterWell Association, Worthington, OH.
Morel, F. M. M., 1983, Principles of aquatic chemistry: Wiley Interscience, New York, NY.
Morgan, J. J., 1967, Application and limitations of chemical thermodynamics in water systems: in Equilibrium Concepts in Natural Water Systems, Advances in Chemistry Series No. 67, American Chemical Society, Washington, DC.
Moses, C. O. and J. S. Herman, 1986, Computer notes-WATIN-A computer program for generating input files for WATEQF: Ground Water, v. 24, pp. 83-89.
Murarka, I. P. and D. A. McIntosh, 1987, Solid-waste environmental studies (SWES): description, status, and available results: EPRI EA-5322-SR, Electric Power Research Institute, Palo Alto, CA.
Neuzil. C. E.,1986, Groundwaterflow in low-permeability environments: Water Resources Research, v. 22, pp. 1163-1195.
Nkedi-Kizza, P., P. S. C. Rao, and A. G. Hornsby, 1985, Influence of organic cosolvents on sorption of hydrophobic organic chemicals by soils: Environmental Science and Technology, v. 19 , pp. 975-979.
Palmer, C. D., 1990, Hydrogeochemistry of the subsurface: in Chemistry of Ground Water, C. Palmer, Editor, Lewis Publishers, Boca Raton, FL (In Review).
Palmer, C. D., and R. L. Johnson, 1989a, Physical processes controlling the transport of contaminants in the aqueous phase: in Transport and Fate of Contaminants in the Subsurface, U.S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/ 625/4-89/019.
Palmer, C. D., and R. L. Johnson, 1989b, Physical processes controlling the transport of non-aqueous phase liquids in the subsurface: in Transport and Fate of Contaminants in the Subsurface, U.S. Environmental Protection Agency, Centerfor Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/ 625/4-89/019.
Paul, E.A. and F.E. Clark, 1989, Soil microbiology and biochemistry: Academic Press, Inc., San Diego, CA.
Perdue, E.M., 1985, Acidic functional groups of humic substances: in Humic Substances in Soil, Sediment, and Water, G.R.Aiken, D.M. McKnight, R.L.Wershaw, and P. MacCarthy, Editors, Wiley Interscience, New York, NY.
Perdue, E. M. and C. R. Lytle, 1983, A distribution model for binding of protons and metal ions by humic substances: Environmental Science and Technology, v. 17, pp. 654-660.
Puri, S., 1984, Aquifer studies using flow simulation: Ground Water, v. 22, pp. 538-543.
Reichenburg, D., 1966, Ion exchange selectivity: in Ion Exchange, Vol. 1, J. A. Marinsky, Editor, Marcel Dekker, New York, NY.
Reuter, J. G., J. J. McCarthy, and E. J. Carpenter, 1979, The toxic effect of copperon occillatoria (Trichodesmium) theibautii: Limnology and Oceanography, v. 24, no. 3, pp. 558-561.
Saffman, P. G. and G. Taylor, 1958, The penetration of a fluid into a porous medium or hele-shaw cell containing a more viscous liquid: Proceeding of the Royal Society of London Series A, v. 245, pp. 312-329.
Scheigg, H. 0.,1984, Considerations on water, oil, and air in porous media: Water Science Technology, v. 17, pp. 467-476.
Schindler, P. W., B. Furst, R. Dick, and P. U. Wolf, 1976, Ligand properties of surface silanol groups. I. Surface complex formation with Fe3+, Cu2+, and Pb2+: Journal of Colloid and Interface Science, v. 55, no. 2, pp. 469-475.
Schnitzer, M., 1969, Reactions between fulvic acid, a soil humic compound, and inorganic soil constituents: Soil Science of America Proceedings, v. 33, pp. 75-81.
Schwarzenbach, R. and J. Westall, 1981, Transport of nonpolar organic compounds from surface water to groundwater: laboratory sorption studies: Environmental Science and Technology, v. 15, pp. 1360-1367.
Schwille, F.,1988, Dense chlorinated solvents in porous and fractured media: model experiments: J. F. Pankow, Translator. Lewis Publishers, Chelsea, MI.
Shelton, M. L., 1982, Ground-water management in basalts: Ground Water, v. 20, pp. 86-93.
Sholkovitz, E. R., 1985, Redox-related geochemistry in lakes: alkali metals, alkaline earth metals, and cesium-137: in Chemical Processes in Lakes, W. Stumm, Editor, Wiley-Interscience, New York, NY.
Siegrist, H. and P. L. McCarty, 1987, Column methodologies for determining sorption and biotransformation potential for chlorinated aliphatic compounds in aquifers: Journal of Contaminant Hydrology, v. 2, pp. 31-50.
Sims, J. L., R. C. Sims, and J. E. Matthews, 1990, Approach to bioremediation of contaminated soil: Hazardous Waste & Hazardous Materials, v. 7, pp.117-149.
Sims, R. C., D. L. Sorensen, J. L. Sims, J. E. McLean, R. Mahmood, and R. R. Dupont, 1984, Review of In-Place Treatment Techniques for Contaminated Surface Soils, Volume 2: Background Information for In Situ Treatment, U. S. Environmental Protection Agency, Hazardous Waste Engineering Research Laboratory, Cincinnati, OH, EPA/540/2-84-003a.
Smith, R. M. and A. E. Martell, 1975, Critical stability constants, vol. 2: Amines, Plenum Press, New York, NY.
Sposito, G., 1984, The surface chemistry of soils: Oxford University Press, New York, NY.
Sposito, G., 1986, Sorption of trace metals by humic materials in soils and natural waters: CRC Critical Reviews in Environmental Control, v. 16, pp. 193-229.
Stevenson, F. J., 1982, Humus chemistry: genesis, compositions, reactions: Wiley Interscience, NewYork, NY.
Strecker, E. W. and W. Chu, 1986, Parameter identification of a ground-water contaminant transport model: Ground Water, v. 24, pp. 56-62.
Stumm, W. and J. J. Morgan, 1981, Aquatic chemistry, second edition: Wiley Interscience, New York, NY.
Stu mm, W., H. Hohl, and F. Dalang,1976, Interaction of metal ions with hydrous oxide surfaces: Croatica Chemica Acta, v. 48, no. 4, pp. 491-504.
Suflita, J. M., 1989a, Microbial ecology and pollutant biodegradation in subsurface ecosystems: in Transport and Fate of Contaminants in the Subsurface, U.S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/625/4-89/019.
Suflita, J. M., 1989b, Microbiological principles influencing the biorestoration of aquifers: in Transport and Fate of Contaminants in the Subsurface, U.S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/625/4-89/019.
Tanford, C., 1973, The hydrophobic effect: formation of micelles and biological membranes: Wiley and Sons, New York, NY.
Thurman, E. M.,1985, Humid substances in the ground water: in Humid Substances in Soil, Sediment, and Water: Geochemistry, Isolation, and Characterization, G. R. Aiken, D. M. McKnight, R. L. Wershaw, and P. MacCarthy, Editors, Wiley Interscience, New York, NY.
Updegraff, D. M., 1982, Plugging and penetration of petroleum reservoir rock by microorganisms: Proceedings of 1982 International Conference on Microbial Enhancement of Oil Recovery, May 16-21, Shangri-La, Afton, OK.
U. S. Environmental Protection Agency, 1985, Protection of public water supplies from ground-water contamination: U. S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH.
U. S. Environmental Protection Agency, 1989, Transport and fate of contaminants in the subsurface: EPA/625/489/019, U.S. Environmental Protection Agency, Center for Environmental Research Information, Cincinnati, OH, and Robert S. Kerr Environmental Research Laboratory, Ada, OK.
van der Heidje, P. K. M., 1984a, Availability and applicability of numerical models for ground water resources management: IGWMC Report No. GWMI 84-19, International Ground Water Modeling Center, Holcolm Research Institute, Butler University, Indianapolis, IN.
van der Heidje, P. K. M.,1984b, Utilization of models as analytic tools for groundwater management: IGWMC Report No. GWMI 84-18, International Ground Water Modeling Center, Holcolm Research Institute, Butler University, Indianapolis, IN.
van der Heidje, P. K. M. and others, 1985, Groundwater management: the use of numerical models, second edition: AGU Water Resources Monograph No. 5, American Geophysical Union, Washington, DC.
van der Heidje, P.K.M. and P. Srinivasan, 1983, Aspects of the use of graphic techiniques in ground-water modeling. IGWMC Report No. GWMI 83-11, International Ground Water Modeling Center, Holcolm Research Institute, Butler University, Indianapolis, IN.
Waksman, S. A., 1916, Bacterial numbers in soil, at different depths, and in different seasons of the year: Soil Science, v. 1, pp. 363-380.
Weaver, J., C. G. Enfield, S. Yates, D. Kreamer, and D. White, 1989, Predicting subsurface contaminant transport and transformation: considerations for model selection and field validation: U. S. Environmental Protection Agency, Robert S. Kerr Environmental Research Laboratory, Ada, OK, EPA/600/2-89/045.
Westall, J. C., J. L. Zachary, and F. M. M. Morel, 1976, MINEQL: a computer program for the calculation of chemical equilibrium composition of aqueous systems: Technical Note No. 18, Massachusetts Institute of Technology, Boston, MA.
Williams, P. A., 1985, Secondary minerals: natural ion buffers: in Environmental Inorganic Chemistry, K. J. Irgolic and A. E. Martell, Editors, VCH Publishers, Deerfield Beach, FL.
Zachara, J. M., and others, 1988, Influence of cosolvents on quinoline sorption by subsurface materials and clays: Journal of Contaminant Hydrology, v. 2, pp
